2019-08-19
8月13日,国际学术期刊Cell Discovery 在线发表了中国科学院上海营养与健康研究所中科院计算生物学重点实验室(马普计算生物学研究所)邵振课题组研究论文“MAP: model-based analysis of proteomic data to detect proteins with significant abundance changes”,报道了一种新计算模型MAP,用于统计分析基于同位素标记产生的定量蛋白质组数据并鉴定其中差异表达的蛋白质。 基于同位素标记和质谱技术的定量蛋白质组实验(如iTRAQ、TMT和SILAC等)能同时检测数千甚至上万个蛋白质在不同样本之间的相对丰度或表达差异。这类数据已有的差异表达分析方法大多依赖于对并行或已有的技术重复数据进行前期比较来构建实验的技术误差模型,并以它为基础检验每个蛋白质在被比较样本之间表达差异的统计显著性。该方法占用了有限的实验通道,也难以保证误差模型的精确适用性。 针对这一局限,在MAP模型中研究人员发展了一种新颖的分步回归(step-by-step regression)分析流程,实现直接对被比较的两个iTRAQ样本构建技术误差模型。在此类研究中,一个常用的经验假设是技术误差对样本间每个蛋白质iTRAQ信号log2比率(log2-ratio)的贡献服从以0为中心的正态分布N(0, σ2)。其中,方差σ2依赖于该蛋白质的信号强度,并且常被用一个指数衰减函数来刻画其依赖关系,即所要构建的全局误差函数。MAP模型首先使用滑动窗口扫描两个样本的M-A图,同时对窗口中0附近的log2比率进行线性建模,以其斜率的平方作为误差函数的局域估计。然后,对所得局域估计进行第二轮指数拟合,获得被比较样本的全局误差函数,并以它为参照计算每个蛋白质信号差异的显著性P值(图1)。关闭窗口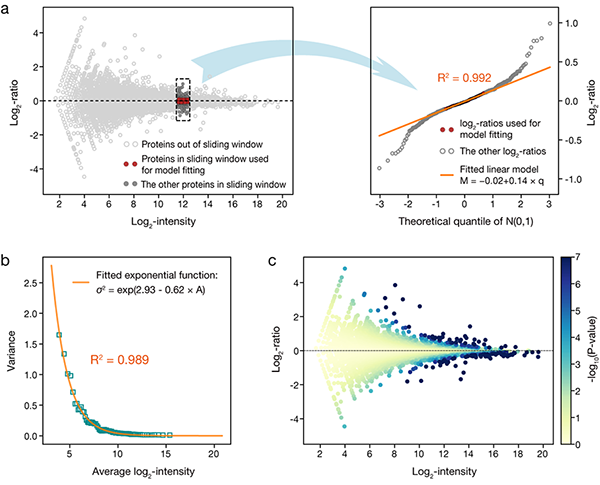
图1:MAP模型的分步回归分析流程:(a)局域线性拟合;(b)全局指数拟合构建技术误差模型;(c)计算每个蛋白质信号差异的显著性P值。 同位素标记定量蛋白质组数据长期存在比率压缩的难题。研究人员使用MAP模型分别比较分析了三个批次产生的小鼠胚胎干细胞分化前后蛋白质组数据,发现蛋白质iTRAQ信号log2比率在不同批次间关联很低(图2a),可能是因为技术误差对其贡献所服从的正态分布N(0, σ2)在批次间各不相同。根据MAP模型,研究人员提出使用每个批次的全局误差函数对其中每个蛋白质iTRAQ信号的log2比率进行重标度(rescaling),使得在不同批次中技术误差对其贡献均服从标准正态分布N(0, 1),从而发展了一个新的Z统计量。比较不同批次蛋白质Z统计量之间的关联,可以发现它具有明显更好的可重复性(图2b)。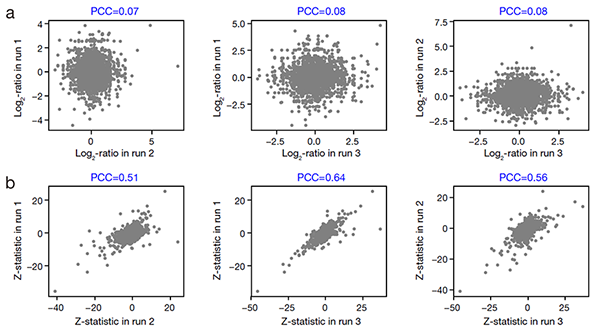
图2:不同批次蛋白质iTRAQ信号的log2比率(a)和Z统计量(b)的皮尔森关联系数。 为方便蛋白质组领域研究者使用MAP模型进行数据分析,研究人员搭建了一个网络服务平台(http://bioinfo.sibs.ac.cn/shaolab/MAP)。该平台额外搭载了一个整合分析模块,能够通过整合多个批次生物重复比较结果的次优P值或者平均Z统计量来最终鉴定差异表达蛋白质,并新发展了一种分析方法用于估测基于这些统计量所定义差异表达蛋白质的错误发现率(FDR)。此外,在用于双样本比较的MAP模型基础上,研究人员还通过分别用样本方差和卡方分布分位数取代原分步回归建模流程中所使用的log2比率和标准正态分布分位数,进一步发展了适用于多样本比较的拓展eMAP模型。 上述研究由营养与健康所研究助理李木山和博士研究生涂世奇等在研究员邵振的指导下,与中科院植物生理生态研究所、复旦大学上海医学院和美国西南医学中心等多家单位的研究人员合作完成,得到国家自然科学基金委、科技部和中科院等的资助。 论文链接

